Machine learning links material composition and performance in catalysts
Understanding how to design better catalysts could enable sustainable energy tech and make everyday chemicals more environmentally friendly.
Understanding how to design better catalysts could enable sustainable energy tech and make everyday chemicals more environmentally friendly.
In a finding that could help pave the way toward cleaner fuels and a more sustainable chemical industry, researchers at the University of Michigan have used machine learning to predict how the compositions of metal alloys and metal oxides affect their electronic structures. The electronic structure is key to understanding how the material will perform as a mediator, or catalyst, of chemical reactions.
“We’re learning to identify the fingerprints of materials and connect them with the material’s performance,” said Bryan Goldsmith, the Dow Corning Assistant Professor of Chemical Engineering.
A better ability to predict which metal and metal oxide compositions are best for guiding which reactions could improve large-scale chemical processes such as hydrogen production, production of other fuels and fertilizers, and manufacturing of household chemicals such as dish soap.
“The objective of our research is to develop predictive models that will connect the geometry of a catalyst to its performance. Such models are central for the design of new catalysts for critical chemical transformations,” said Suljo Linic, Martin Lewis Perl Collegiate Professor of Chemical Engineering.
One of the main approaches to predicting how a material will behave as a potential mediator of a chemical reaction is to analyze its electronic structure, specifically the density of states.This describes how many quantum states are available to the electrons in the reacting molecules and the energies of those states.
Usually, the electronic density of states is described with summary statistics—an average energy or a skew that reveals whether more electronic states are above or below the average, and so on.
“That’s okay, but those are just simple statistics. You might miss something. With principal component analysis, you just take in everything and find what’s important. You’re not just throwing away information,” said Goldsmith.
Principal component analysis is a classic machine learning method, taught in introductory data science courses. They used the electronic density of states as input for the model, as the density of states is a good predictor for how a catalyst’s surface will adsorb, or bond with, atoms and molecules that serve as reactants. The model links the density of states with the composition of the material.
Unlike conventional machine learning, which is essentially a black box that inputs data and offers predictions in return, the team made an algorithm that they could understand.
“We can see systematically what is changing in the density of states and correlate that with geometric properties of the material,” said Jacques Esterhuizen, a PhD student in chemical engineering and first author on the paper in Chem Catalysis.
This information helps chemical engineers design metal alloys to get the density of states that they want for mediating a chemical reaction. The model accurately reflected correlations already observed between a material’s composition and its density of states, as well as turning up new potential trends to be explored.
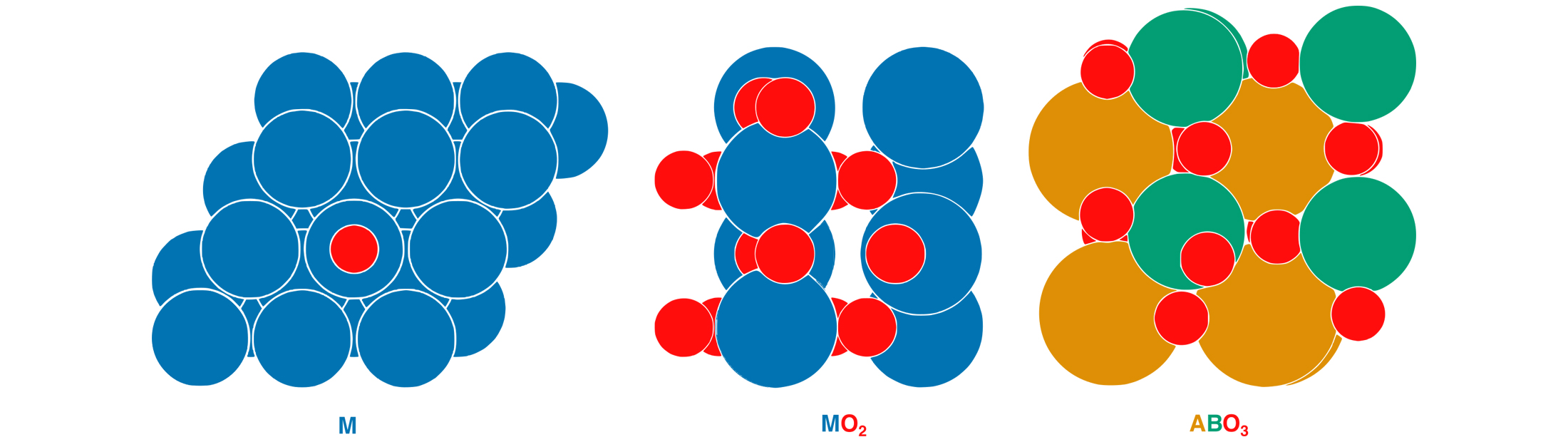
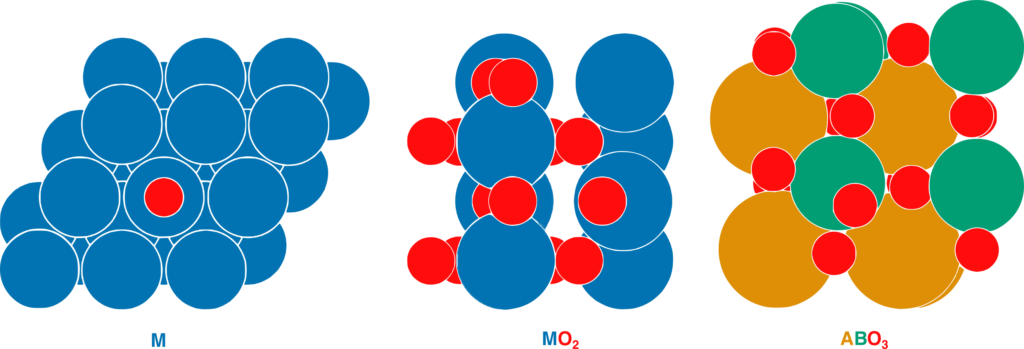
The model simplifies the density of states into two pieces, or principal components. One piece essentially covers how the atoms of the metal fit together. In a layered metal alloy, this includes whether the subsurface metal is pulling the surface atoms apart or squeezing them together, and the number of electrons that the subsurface metal contributes to bonding. The other piece is just the number of electrons that the surface metal atoms can contribute to bonding. From these two principal components, they can reconstruct the density of states in the material.
This concept also works for the reactivity of metal oxides. In this case, the concern is the ability of oxygen to interact with atoms and molecules, which is related to how stable the surface oxygen is. Stable surface oxygens are less likely to react, whereas unstable surface oxygens are more reactive. The model accurately captured the oxygen stability in metal oxides and perovskites, a class of metal oxides.
The study is titled, “Uncovering electronic and geometric descriptors of chemical activity for metal alloys and oxides using unsupervised machine learning.” This work was supported by the Department of Energy and the University of Michigan.
Uncovering electronic and geometric descriptors of chemical activity for metal alloys and oxides using unsupervised machine learning.